최신 단백질 구조 예측 모델인 AlphaFold3(AF3)는 혁신적인 정확도를 보여주지만, 여전히 원자 수준의 미세한 steric clash(원자 간 충돌)나 기하학적 왜곡(Geometry outliers) 문제를 완벽히 해결하지 못하는 경우가 많습니다.
본 분석에서는 EGFR 구조를 대상으로 기본 AF3 모델과 이를 hanjarifold 엔진으로 정밀화(Refinement)한 모델의 MolProbity 데이터를 비교 검증하였습니다. 그 결과, hanjarifold가 단백질 구조의 물리적 타당성을 어디까지 끌어올릴 수 있는지 확인했습니다.
1. 전 세계 상위 1%의 정밀도: MolProbity Score 및 Percentile
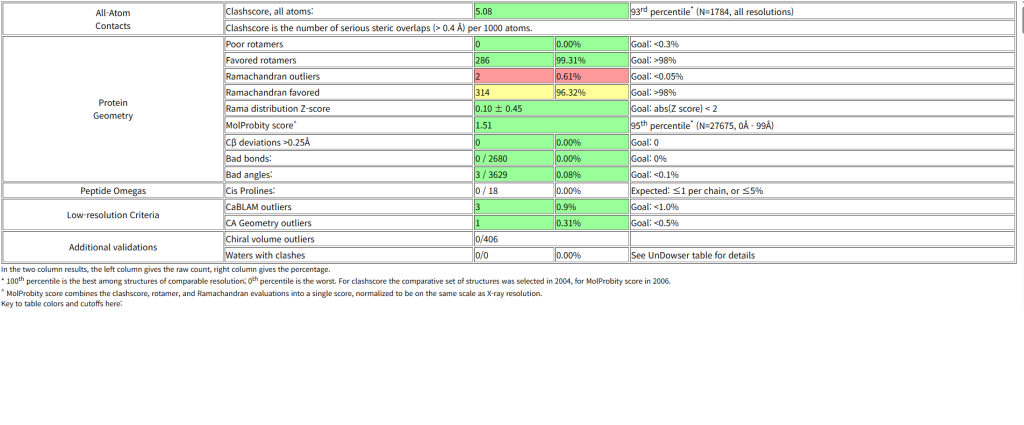
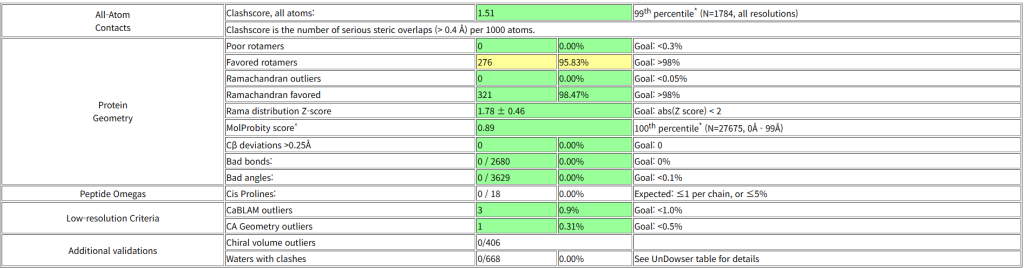
구조의 전체적인 품질을 대변하는 MolProbity Score에서 압도적인 차이가 확인되었습니다.
| 평가 항목 | EGFR_AF3 (기본) | EGFR_AF3_hanjarifold | 비고 |
| MolProbity Score | 1.51 | 0.89 | hanjarifold 압승 |
| Percentile | 95th | 100th | 전 세계 최상위 수준 |
기본 모델도 95th percentile로 우수한 편이지만, hanjarifold는 0.89점이라는 경이로운 점수를 기록하며 100th percentile(상위 0% 수준)에 도달했습니다. 이는 실험적으로 규명된 초고해상도 결정 구조와 비교해도 손색없는 물리적 신뢰성을 확보했음을 의미합니다.
2. Steric Clash의 완벽한 제거: Clashscore 분석
원자 간의 비정상적인 겹침을 나타내는 Clashscore는 시뮬레이션이나 도킹 계산 시 노이즈를 발생시키는 주요 원인입니다.
-
EGFR_AF3: 5.08 (93rd percentile)
-
EGFR_AF3_hanjarifold: 1.51 (99th percentile)
hanjarifold는 기본 모델 대비 충돌 지수를 약 70% 이상 감소시켰습니다. 원자 배치를 최적화하여 분자 내부의 에너지 상태를 가장 안정적인 형태로 재구현했음을 보여줍니다.
3. 기하학적 결함 제로(Zero Defect) 달성
단백질의 backbone과 side-chain이 가지는 물리적 제약 조건을 완벽하게 충족했습니다.
-
Ramachandran Outliers: 기본 모델의 이상치(0.61%)를 0.00%로 완전히 교정하여 모든 잔기를 허용 영역 안으로 배치했습니다.
-
Bad Bonds & Angles: 기본 모델에서 발견된 3개의 Bad angles를 포함하여, 모든 결합 길이와 각도에서의 오류를 0개(0.00%)로 정정했습니다.
-
Rotamer Integrity: 선호되는 로타머(Favored rotamers)의 비율을 최적화하면서도 이상치는 단 하나도 허용하지 않는 정밀함을 보였습니다.
4. 고해상도 분석을 위한 최적의 모델
저해상도 기준인 CaBLAM 및 CA Geometry 수치 역시 목표치 이내에서 안정적으로 관리되고 있으며, 특히 Chiral volume outliers가 전혀 없는 깨끗한 데이터를 유지하고 있습니다. 이는 hanjarifold가 단순히 좌표를 조정하는 것을 넘어, 생물학적·화학적 원리를 엄격히 준수하며 구조를 재구성한다는 증거입니다.
결론: 왜 hanjarifold인가?
이번 비교 분석을 통해 EGFR_AF3_hanjarifold 모델은 단순한 예측 모델을 넘어 ‘실행 가능한(Actionable) 고정밀 구조’임을 입증했습니다.
-
극한의 물리적 타당성: 이상 각도와 충돌이 제거되어 분자 역학(MD) 시뮬레이션 시 평형화 시간을 단축하고 계산 오류를 방지합니다.
-
신뢰도 높은 도킹 시뮬레이션: Side-chain 정밀도가 극대화되어 약물 후보 물질과의 결합 에너지를 더욱 정확하게 계산할 수 있습니다.
-
구조 생물학적 완성도: 전 세계 구조 데이터베이스 기준 상위 1% 내외의 품질을 달성하여, 논문 게재 및 학술적 근거로서의 가치를 확보했습니다.
정밀한 단백질 설계와 분석이 필요한 연구자에게 hanjarifold는 선택이 아닌 필수적인 정밀화 과정이 될 것입니다.
답글 남기기
댓글을 달기 위해서는 로그인해야합니다.